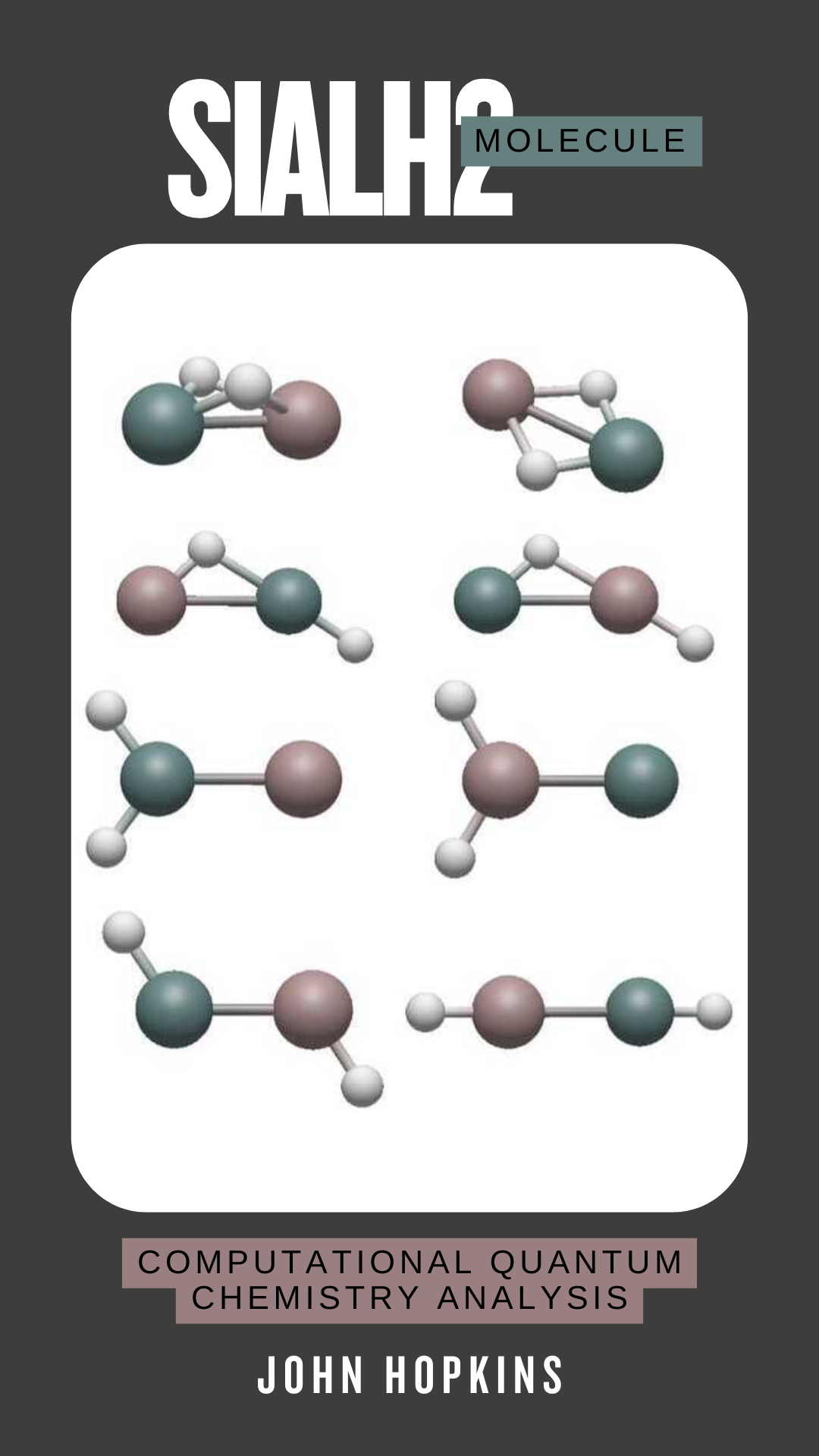
SiAlH2 Molecule Structure Analysis
Awardee: John Hopkins
Award Year: 2025
Understanding the structure of group 13 and 14 element-containing M2H2 acetylene analogues has been of interest to ab-initio computational chemistry for some time. However, analysis of the possible structures of acetylene analogues that contain two different elements, especially elements from two different chemical groups, has not been widely seen in the literature. In this study, we evaluate the geometric and energetic properties of seven isomers for the SiAlH2 molecule by performing harmonic analysis to discern whether these structures are energy minimum or transition states, depending on the presence of imaginary frequencies. Computations are performed using state-of-the-art ab-initio methods in the CFOUR (Coupled-Cluster techniques for Computational Chemistry) program up to coupled-cluster with single, double, and perturbative triples [CCSD(T)] levels of theory to optimize geometric parameters for the structures. The results of this study can be used to aid future experimental work with SiAlH2 and similar molecules, especially acetylene-like molecules containing mixed atoms from the carbon and boron groups.